By YAN Fusheng (Staff Reporter)
In the July 9 issue of Cell, a group of scientists presented a general platform to create covalent protein drugs from various protein-protein interactions by incorporating an unnatural amino acid into one of the proteins, which can then target the other in vitro, on cell surfaces, and in mice. As a proof of concept, they chose the binding pair of PD-1/PD-L1, a typical ‘loophole’ that tumor cells can exploit to evade immune detection and elimination; therefore, the blockage of this coupling is therapeutically relevant. By expanding functionality at a picked amino acid in PD-1, they engineered PD-1 into a covalent binder, which greatly revives or enhances the anti-tumor immunity of T cells in immune-humanized mice by binding covalently to PD-L1.
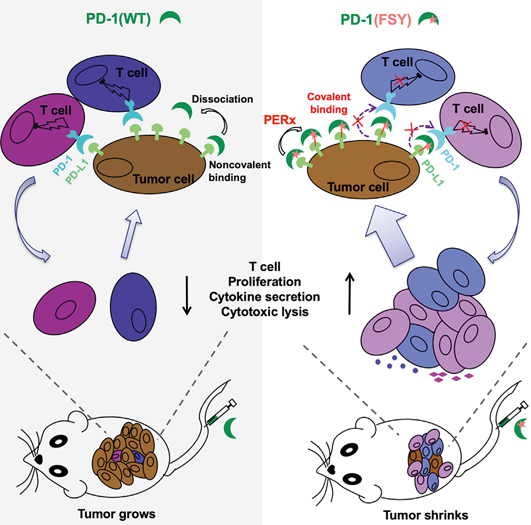
Engineering PD-1 into a covalent drug. Solid tumor cell surface is rich in PD-L1 proteins that help the tumor cells hiding from immune attack via the binding pair of PD-1/PD-L1. The blockage of this coupling is therefore therapeutically relevant. Left: The injection of wildtype PD-1 proteins fails to halt tumor growth due to the constant dissociation of PD-1 from PD-L1. Right: PD-1(FSY) with expanded functionality that enables it to forge a covalent bond towards PD-L1 in proximity, effectively shrinks the tumor in mice. (Image by WANG’s lab)
One major purpose of studying how things work within a cell or a body is to come up with a trouble-shooting so that we can fix the breakdowns by rewiring pieces around, and kick the system back to normal.
This is particularly truthful in studying tumor cells where something must be out of order. Usually, we have the immune system on our side that acts to get rid of unhealthy and ailing cells. However, tumor cells are mischiefs, good at tapping the right buttons to evade immune surveillance and elimination.
Tap the Buttons
Solid tumor cell surface is rich in PD-L1 proteins that help the tumor cells hiding from immune attack through binding to PD-1 (a receptor on T cell that regulates T cell activity). The coupling of PD-1/PD-L1 acts as a type of ‘off-switch’ and thereby suppresses T cell’s anti-tumor immunity (Figure 1). The blockage of this coupling is hence a hot pursuit by big pharms, and several monoclonal antibodies target this coupling have been developed to revive anti-tumor immunity and kill the tumor cells. Though surprising good therapeutic outcomes have been seen in many cases, the response varies in most patients. Antibodies have inherent limitations including poor tissue/tumor penetrance due to the large size, and adverse side effects that could deplete immune cells and induce tissue damage.
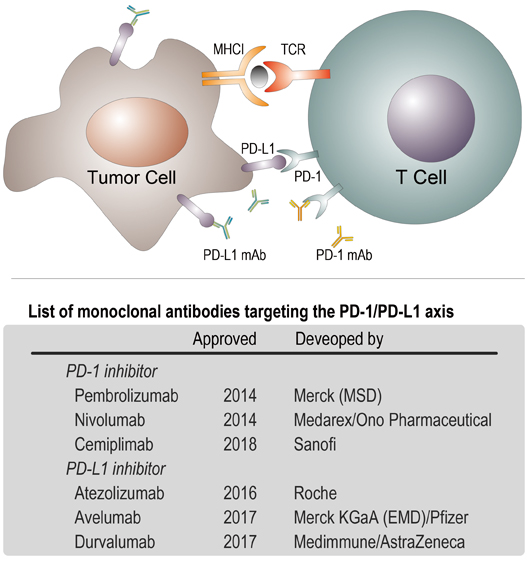
Figure 1. Up: PD-1 is a checkpoint protein on immune cells called T cells. It normally acts as a type of ‘off switch’ that helps keep the T cells from attacking other cells in the body. It does this when it attaches to PD-L1, a protein on some normal (or cancerous) cells. When PD-1 binds to PD-L1, it basically tells the T cell to leave the other cell alone, which is the reason to be called ‘checkpoint’. Solid tumor cell surface is rich in PD-L1 proteins that help tumor cells avoid immune attacks; Down: a list of commercial monoclonal antibodies targeting to the binding pair of PD-1/PD-L1. (Credit: YAN Fusheng)
One alternative is by using mutants of high-affinity PD-1 that are relatively small in size. However, these molecules failed to yield better results, and still required an extra appendage to extend their in vivo half-life to ensure therapeutic effects; otherwise, they will be quickly removed out of the circulation and ended up in piss.
Though these PD-1 mutants are both small-size and superior in binding PD-L1 on tumor cells, the researchers pondered that the Achilles’s heel for these molecules is the reversible nature of noncovalent bindings: PD-1 will constantly detach from PD-L1 and fail to sustain a therapeutic effect.
The forces that keep proteins binding together are relative week, and once a while the binding pair will detach from each other. When a drug detaches from its target, its therapeutic effect is likely to suspend. To make up what causes by the constant detachment, one method is keeping the drug concentration relatively high around the targets, so that another drug would quickly take over the shift. This demands either a higher dosing or a longer half-life in vivo coupled with tumor-targeted drug accumulation: the former would probably cause adverse side effect and the latter is clinically hard to achieve.
Switch ON Elegantly
To bypass these two challenges and overcome the constant detachment, a research team led by WANG Qian from the Hangzhou Research Institute of Technical Institute of Physics and Chemistry, the Chinese Academy of Sciences (CAS) and his collaborators presented a method to merge PD-1 and PD-L1 covalently in vivo that possesses infinite affinity.
The first challenge to make this happen is that two native proteins won’t spontaneously form irreversible bonds, which is written in the chemical nature of the functionalities in the 20 essential amino acids that build up any protein. Though there are many routine ways to introduce external chemical groups to a protein, it is quite hard to place an external chemical widget to a precise site in a protein. The second challenge is that the intended reaction may not discriminate a protein target from innocent bystanders, which may exhaust the drug before it reaches its target and might also cause adverse side effects.
The most elegant and controllable way to site-specifically incorporate external functionalities in a protein is during the actual protein translation – a process of synthesizing protein through reading out the triple codes written in the message RNA – using the so-called ‘genetic code expansion’. The core of genetic code expansion is to introduce an unnatural amino acid (Uaa) to a precise location in a protein. Its typical workflow is illustrated in Figure 2.
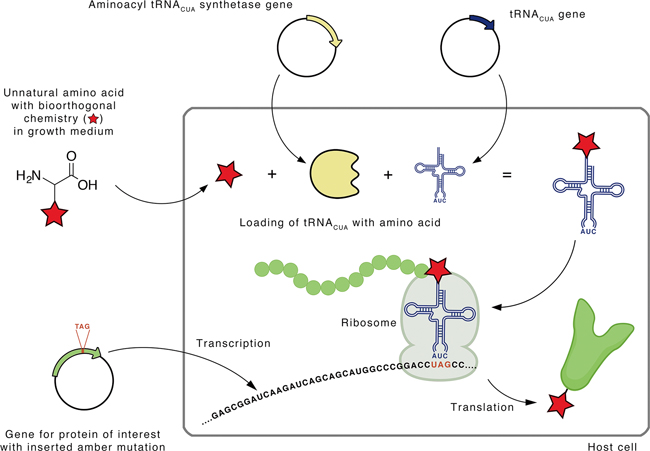
Figure 2. Site-specific incorporation of an unnatural amino acid into a protein by an expanded genetic code. A mutant aminoacyl-tRNACUA synthetase recognizes the unnatural amino acid bearing the desired functionality and loads it to the corresponding tRNACUA to form the Uaa-tRNACUA. Using the native protein-synthesizing machinery, the loaded Uaa is incorporated at the desired site of the protein via tRNACUA-mediated recognition of the amber codon, the triple of ‘UAG’. (Image by E. Redeker)
Genetic code expansion is based on two assembly lines of biorthogonal chemistry –chemical reactions that can occur inside a cell without interfering with native biochemical processes. The first line of biorthogonality acts to load Uaa to the transfer RNA (Uaa-tRNACUA) without any promiscuity to the native processes of loading 20 essential natural amino acids to their respective tRNAs. The second line of biorthogonality aims to re-allocate the amber codon – the triplet of ‘UAG’ that is natively read as a ‘stop signal’ to terminate protein synthesis – to encode the Uaa, and thereby insert an artificial functionality carried by the Uaa to a specific site in the protein.
Lay the Foundation
The research team formerly engineered a genetically expanded system to incorporate unnatural amino acids (Uaas) into proteins to react with target natural amino acids via proximity-enabled reactivity (Figure 3). They designed the first latent bioreactive Uaa named fluorosulfate-L-tyrosine (FSY) that can remain inert towards free amino acids and specifically react with three natural amino acids – lysine (K), histidine (H) and tyrosine(Y) – in proximity on proteins in vivo via sulfur–fluoride exchange (SuFEx) chemistry. Notably, FSY can be introduced into proteins in E. coli or mammalian cells.
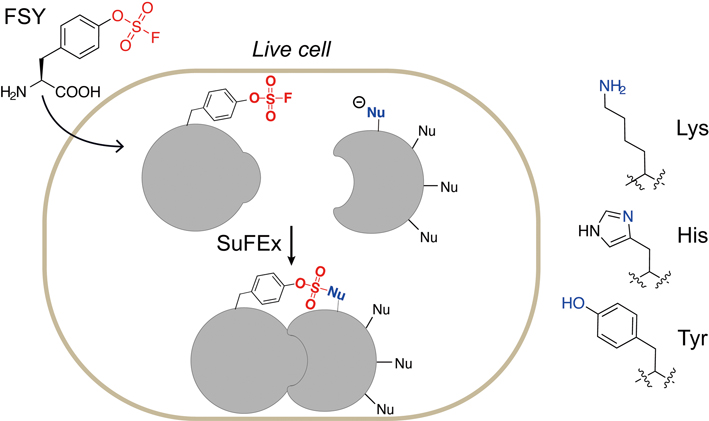
Figure 3. An unnatural amino acid, fluorosulfate-l-tyrosine (FSY), can be incorporated into proteins via an expanded genetic code. Incorporated FSY selectively reacts with lysine, histidine and tyrosine residues (all of them have an electron-donating functionality) in close proximity, forming a covalent bond. (Image by WANG’s lab)
Forge the Bond
In this work, the researchers used the E. coli platform to produce FSY-incorporated PD-1, in which they carefully pick three sites (Q75, D77 and A129) and replace each one individually with the latent bioreactive FSY (Figure 4). These picked sites are all in proximity with a target natural amino acid [histidine69 (H69) or lysine124 (K124)] at the binding interface on the PD-L1 protein (Figure 4C).
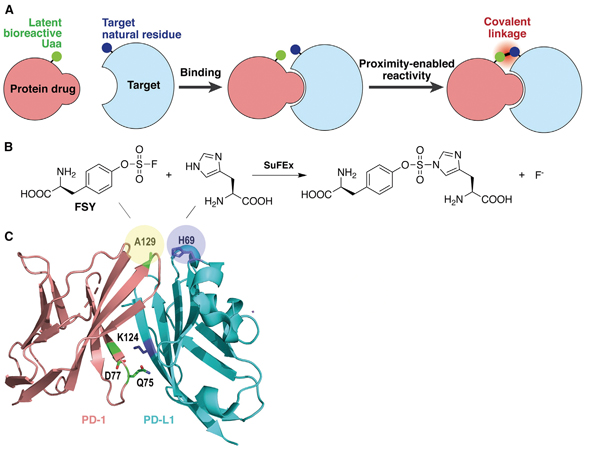
Figure 4. Developing covalent protein drugs via proximity-enabled click chemistry. (A) The latent bioreactive Uaa forms an irreversible bond with the target natural amino acid in proximity. (B) FSY reacts with a proximal histidine via click chemistry. (C) Crystal structure of human PD-1/PD-L1 complex shows the three selected sites (shadowed in yellow) on PD-1 for FSY replacement. (Image by WANG’s lab)
By examining the band-shifting mode of PD-L1 via SDS-PAGE (a gel electrophoresis technique that separate proteins by size into different bands) coupled with immunoblotting (an antibody-based method to visualize the bands of a target protein in gels), they demonstrated that human PD-1(FSY), other than the wildtype PD-1, covalently bound to human PD-L1. The covalent binding can be achieved efficiently and specifically with purified target proteins and target proteins expressed on cultured cancer cells in vitro. Notably, a mutation of PD-L1 at His69 abolished the covalent binding of human PD-1(FSY), indicating that human PD-1(FSY) covalently targeted human PD-L1 with high specificity.
Then they further tested whether PD-1(FSY) could covalently bond with human PD-L1 in tumor tissue in vivo. They picked immunodeficient mice to engraft human cancer cells to form solid tumors and let it grow to size in the animals. The PD-1(WT, wildtype) and PD-1(FSY) was introduced into mice via tail intravenous injection or peri-tumoral injection. After 3 hours, the tumors were dissected for immunoblotting of PD-L1. PD-1(WT) again did not yield any crosslinking with PD-L1, whereas PD-1(FSY) showed apparent covalent crosslinking with PD-L1 in both types of tumor and through both injection methods, with the crosslinking efficiency increased with the doses of PD-1(FSY) injected and target proteins expressed in tumor tissues in vivo.
Taken together, human PD-1(FSY) covalently bound to human PD-L1 through FSY129 of PD-1 reacting with His69 of PD-L1 in vitro and in vivo.
Have the Edge
Then they sought to evaluate the anti-tumor effect of PD-1(FSY), the first proof-of-concept covalent protein drug, in reviving the dampened T cell activity. Once again, they resorted to immune deficient mouse models that can take in externally infused human immune cells without incurring rejection. This allows the researchers to evaluate the engrafted human immune system against the engrafted human tumors in mice. They found that PD-1(FSY) reached the same antitumor effect with atezolizuma (an FDA-approved cancer treating monoclonal antibody against human PD-L1), while the PD-1(WT) lag behind.
They further evaluated if PD-1(FSY) could improve the therapeutic potential of CAR-T cells (short for chimeric antigen receptor T cells, genetically engineered T cells that target tumor cells with a certain tumor antigen on their surface) in treating human solid tumors. CAR-T cells have shown an outstanding record in treating hematopoietic malignancies, but are not effective against solid tumors due to the presence of tumor immune suppressive microenvironment, which necessitates the need to develop methods to enhance CAR-T activity in solid tumors.
They used a type of CAR-T cells that are engineered to target the human cancer cells used in this study. Their data showed that, in the presence of target human cancer cells, PD-1(FSY) can significantly enhance CAR-T cells activation in vitro, whereas PD-1(WT) can not. Similarly, they found that PD-1(FSY) can achieve much better antitumor effect over PD-1(WT) in vivo, in terms of enhancing CAR-T cell infiltration and activity in the engrafted solid tumors.
To draw a more reliable conclusion, they re-evaluated the therapeutic potential of PD-1(FSY) in an immune-humanized mouse model. Compared to the mouse models mentioned above, the humanized mice (Hu-mice) contains a more comprehensive array of human lymphohematopoietic cells including T cells, B cells, and dendritic cells, allowing many important human immune processes to occur in mice.
In this immune-humanized mouse model, they consistently found that PD-1(FSY) exhibited far better therapeutic efficacy than PD-1(WT), and even better than atezolizumab in reducing tumor volume and weight at the same dose.
They also extended their concept to another pair of proteins and successfully demonstrate the general applications of this covalent drug development strategy.
Apart from opening a novel avenue for developing protein therapeutics, proximity-enabled click chemistry also enables scientists to selectively inhibit or activate signal pathways with high specificity, which makes an important addition to the toolbox for basic biological research and synthetic biology.
References
Q. Li, Q. Chen, P. C. Klauser, M. Li, F. Zheng, N. Wang, X. Li, Q. Zhang, X. Fu, Q. Wang*, Y. Xu*, L. Wang*, (2020) Developing covalent protein
drugs via proximity-enabled reactive therapeutics. Cell 182, 85. doi: 10.1016/j.cell.2020.05.028.
N. Wang, B. Yang, C. Fu, H. Zhu, F. Zheng, T. Kobayashi, J. Liu, S. Li, C. Ma, P. G. Wang, Q. Wang, L. Wang, (2018) Genetically encoding
fluorosulfate-l-tyrosine to react with lysine, histidine, and tyrosine via sufex in proteins in vivo. Journal of the American Chemical Society
140, 4995. doi: 10.1021/jacs.8b01087.
E. Steen Redeker, D. T. Ta, D. Cortens, B. Billen, W. Guedens, P. Adriaensens, (2013) Protein engineering for directed immobilization.
Bioconjugate Chemistry 24, 1761. doi: 10.1021/bc4002823.