— An Interview with Prof. CHEN Lingling

Overshadowed by DNA and their coding counterparts, non-coding RNAs remained in the darkness for a long time as “junk” or “useless” fragments in the genome, until only about a decade ago, when analysis of complete sequencing of human genome brought it into attention. As the afterglow of the ground-breaking Human Genome Program, this has since been a hot frontier of biology, and among those who have been pursuing this genetic “dark matter”, Dr. CHEN Lingling, Principal Investigator working at the Shanghai Institute of Biochemistry and Cell Biology (SIBCB), CAS, has impressed the biological community with her discovery and functional annotation of long non-coding RNAs, and won honors including the Howard Hughes Medical Institute (HHMI) International Research Scholar and the L’Oréal Women in Science. We have the honor to invite her to a conversation with BCAS staff reporter SONG Jianlan at the annual academic conference of the Chinese Society for Cell Biology.

Dr. CHEN Lingling
Winner of the Howard Hughes Medical Institute (HHMI) International Research Scholar,
Recipient of the Young Investigator Award of Chinese Biological Investigators Society (CBIS) and the L’Oréal Women in Science
BCAS: From the very first instance, how did non-coding RNAs, which were thought as redundant sequences, draw the interest from scientists as "dark matter" of the genome?
CHEN Lingling (CHEN): Originally, we thought that human genome mostly consisted of genes, the codes that guide the assembling of proteins, while non-coding fragments accounted for only a small fraction of the total base sequences. This belief did not change until 2001, when the sequencing of the human genome was first published. Biologists were startled by the number of genes: they anticipated around 100,000, only to find less than 35,000 (which was later updated to 21,000) among the 3 billion bases on the strands of our DNAs. What could be happening between genes? This reminded us that something important might have slipped off our attention.
Later analysis of the massive data produced by the complete sequencing of the human genome and transcriptome further surprised many scientists. We used to think that most RNAs were messenger RNAs (mRNAs) transcribed from protein-coding genes on the DNA strand, plus a fraction of RNAs in ribosomes and somewhere else. However, much more RNAs were found. In mice, about 80% of the genome is transcribed into RNAs, and about the same in humans. Initially biologists estimated that a small fraction of the transcriptional RNAs were non-coding, however, it turned out that more than 98% of them do not carry instructions of any protein. Big surprise, right?
According to the “Central Dogma” of biology, genetic information coded by the DNA is transcribed to RNAs and carried out from the nucleus to the cytoplasm to translate into proteins in the cell. It is hard to imagine that in human genome only less than 2% of the transcribed genetic information directly leads to production of proteins. What is the overwhelming majority, the more than 98% of our genome, doing in our body? Could such a massive pool of non-coding sequences mean nothing to our body? This shocking discovery reminded us of the big black hole in our knowledge about our own genome, prompting us to explore and understand the formerly overlooked side of it. Now the regulatory roles played by this “dark matter” of the genome have earned much greater attention from scientists.
BCAS: What do you think we can find out exploring into such subtle sequences?
CHEN: Some diseases remind us that this obscure part of our genome matters a lot. An example is Prader-Willi syndrome (PWS), which stayed off the view of Chinese people until quite lately. It occurs at a rate of about one case in every 20,000 newly born babies. This disease induces very serious subsequences in sufferers, including disordered functioning of pituitary gland and hence disordered secretion of hormones important to physical/mental development. Therefore, the syndromes include slow mental and physical development, intellectual impairment and, very obviously in around 70% cases, obesity. Appalling as the syndromes are, its specific underlying mechanism is still unknown. As early as 40 years ago, scientists found that it is caused by the absence of a sequence from the No. 15 paternal chromosome, a genomic imprinting region that is only expressed paternally, however, no protein-coding gene was found in the most critical sequence involved. In other words, the loss of “junk DNA” has led to the serious disease.

As the afterglow of the ground-breaking Human Genome Program, research in non-coding RNAs brings more and more surprises, subverting the traditional belief of their roles in the genome as “junk sequences.” It loomed big into a hot issue in the early 21st century, and in 2010, Science magazine termed it as the “Dark Matter” in the genome in a special edition featuring “Insights of the Decade.”
Up until now, we do not know how this “dumb” area is functioning in human bodies; hence we do not know exactly what mechanisms underlie the disease. Despite the setback, however, in the past several years we identified two new classes of long noncoding RNA (lncRNA) species termed sno-lncRNAs and SPAs associated with this disease. We found out some epigenetic regulation pathways mediated by the RNAs transcribed from this fragment, therefore providing new insights into the molecular cause of PWS pathogenesis. Nowadays, after years of efforts, this disease has aroused much more concern and attention in China.
So far, evidence has shown that defects in lncRNAs or disorders in their regulations can lead to major diseases, including spinal muscular atrophy, myotonic dystrophy, Alzheimer’s disease, and cancers. Therefore, understanding such lncRNAs will help us solve many downstream issues, including development of new drugs targeting certain loci that regulate related physiological processes.

In a cover article published in Molecular Cell on Oct 26, 2012, CHEN’s lab reported the discovery of a novel class of long noncoding RNAs (lncRNA) that are processed from introns and each capped with an snoRNA on its either end, and revealed its role in the molecular pathogenesis of Prader-Willi Syndrome (PWS). This work was selected as a research highlight by Nature Reviews Molecular Cell Biology, and ranked into “The Best of Molecular Cell 2012” by Cell Press as well. It also caught the eye of the Foundation for Prader-Willi Research. Further in 2016, the same team discovered yet another new class of lncRNA species associated with PWS and reported their discovery as another cover article in Molecular Cell.
Generally speaking, studying lncRNAs might help us understand the more refined regulation of some important physiological processes with new frame of details. Such basic understanding of our own body will lay the foundation for further research aimed at solutions to many issues, including but not limited to understanding the molecular basis of diseases, and contribute to translational medicine and human health.
The exploration into the unknown itself is meaningful due to its fundamental nature. No one knows at what time and in what way it will come to our rescue or find significant applications in our daily life.
BCAS: When did you cast your attention to this area in your academic career? At that time what was the most intriguing question(s) for biologists? What did you first focus on in this area? What did you discover?
CHEN: When I was a graduate student in Gordon Carmichael’s lab at UConn Health Center, I worked on Alu elements, which had been thought as the most abundant “junk” DNA elements and account for over 10% of primate’s genome. I found that inverted repeated Alu elements in the 3’ UTR of mRNAs have the potential to form intramolecular double stranded RNAs (dsRNAs) that act to retain mRNAs in particular nuclear substructures called paraspeckles.
But my attention to lncRNA really came in 2009, when the widespread expression of “mRNA-like” lincRNAs (long intergenic noncoding RNAs) was discovered originating from the regions between genes. In 2009, I worked on the lncRNA called NEAT1 and found that NEAT1 is essential for paraspeckles' integrity and function. Interestingly, the long isoform of it is not polyadenylated, which is seen as a necessary process to produce most known mRNAs and lincRNAs in mature forms. Are all lncRNAs similar to mRNAs? What makes the difference? These interesting questions prompted me to embark on a search for more novel lncRNAs in the nonpolyadenylated transcriptomes. At that time, I had just completed my Ph.D. degree, and had successfully obtained a funding for independent research from the State of Connecticut Stem Cell Grant. I began to develop techniques to visualize and characterize nonpolyadenylated RNAs. The first independent work in my career, this has led to the discovery of several classes of RNA species in my lab at SIBCB, Shanghai.
BCAS: You just mentioned techniques to visualize and characterize nonpolyadenylated RNAs. As we noticed, in your recent work published in Cell this year, you adopted CRISPR/CAS9 technique and super-resolution microscopy to accurately knock out the lncRNA SLERT and precisely locate it in the nucleus. Would you describe how such advanced technology has promoted your research?
CHEN: (Showing the reporter some charts from their experiments) Yes, new technologies definitely greatly speed up the progress of everybody’s research. Look, these are what we can see through a microscope of super-high resolution, the Structure Illumination Microscopy (SIM), whose resolution can reach about 100 nm. We applied SIM in our work published in last May in Cell, to define the ring-like structure with a diameter about 400 nm formed by molecules of protein DDX21. We further demonstrated that these ring-shaped structures are associated with RNA Pol I transcription for production of ribosomal RNAs, and can be modulated by a new lncRNA, named SLERT in the nucleolus. Such an “eye (SIM)” is not sharp enough to hit the atomic level, but good enough in this research to deliver the morphology of these structures. Once we have locked onto a certain area that demands closer observation, we can still take advantage of some atomic-level devices. Indeed, combination of SIM, protein immune-fluorescence and RNA fluorescent in situ hybridization has allowed us to accurately capture morphological details of some proteins and RNAs in a variety of nuclear bodies, as well as to monitor how such structures change in response to some external stimuli, which would otherwise be impossible. Such devices are helping us see what used to be invisible and ultimately better understand them. In addition, with modern technologies like CRISPR/CAS9, we can edit the noncoding sequences of interest more accurately, hence more conveniently to control the output. On the other hand, we are also “seeing” more in a computational sense: combining biochemical and bioinformatics methods, we can concentrate targeted sequences and identify new species of lncRNAs from them. Yeah, introduction of computational techniques has greatly promoted our research.
BCAS: As we noticed, some recent discoveries indicate the “bright side” of non-coding RNAs, revealing their key regulative roles in the cell. For example, in a recent research, your team identified a series of trans-factors regulating circRNA expression, and demonstrated a coordinated regulation of circRNA biogenesis and function by NF90/NF110 in viral infection (Li, et al., Molecular Cell 2017). As we know, in higher eukaryotic organisms, such “dark matter” takes larger a proportion than in lower ones. Do you think this is a coincidence? How would you evaluate the role of such “dark matter” in genomes?
CHEN: Higher proportion of such “dark” sequences could represent better ability of fine-tuned regulations of life activity. The more complicated are the organisms, the more complicated are the physiological processes going on in cells and wider ranges of the body; probably higher organisms conserved such “dark” sequences and developed more sophisticated regulation in their long evolution.
Evidence has shown that non-coding RNAs are potential key regulators in gene expression networks, and exhibit a surprising diversity in shapes and sizes. While exploring in this field, which is still quite new and is thriving into many branches, we need to be very cautious in claiming discoveries or interpreting data. It is still at an early stage, and in the long run a lot of further research might rely on what we are doing today.
BCAS: Last May might mark a milestone in your career life. Not only did you publish one paper in Cell and another in Molecular Cell in the same month, but also you beat about 1,500 rivals and won support from the highly prestigious Howard Hughes Medical Institute (HHMI) International Research Scholar Program. What are you going to study taking this opportunity? Why? What are you expecting from your project?
CHEN: I hope that this funding will enable my lab to recruit more skillful and well-trained postdocs and lab technicians. People means a lot – talented and self-motivated coworkers can inspire more exciting ideas and promote our research.
On the other hand, the new funding will also help us further explore the most fundamental issues in this field that entail long-term efforts. Our team has been studying different types of RNAs using a variety of biochemical, cell biological and genome-wide approaches. In the long run, we aim to understand general rules that govern the processing and functioning of lncRNAs and circRNAs in both health and disease contexts. With this funding, we expect more new discoveries in these directions.
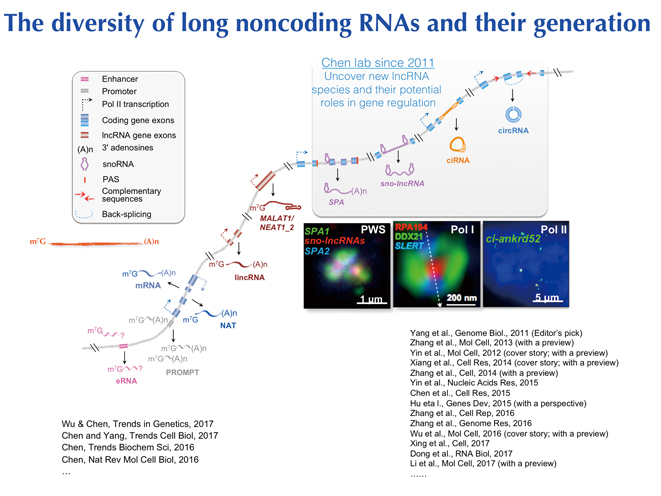
Long non-coding RNAs are emerging as key regulators in gene regulation. CHEN’s lab uncovered many lncRNA species with new formats including broadly expressed circular RNAs and sno-processed lncRNAs over the past few years. In a paper published online in Trends in Genetics on June 17, 2017 (Aug;33(8):540-552), the team gives a review of the diversity of lncRNAs and the underlying mechanisms related to their generation.
BCAS: As the mother of a toddler, and the winner of the L’Oréal Women in Science as well, would you tell us how you balance your research and family?
CHEN: I think it is a matter of time management: you have to work at a very high efficiency and manage to make time for your work. As a frequent traveler, I write papers during my flights; and very often, I get up in small hours to take advantage of the quiet moments when my daughter stays asleep in bed... Thanks to the great support from my family, I am handling this fairly well – so far so good.
Maybe my MBA background helps me a little bit. Running a lab is just like starting up a small business: when you are doing something bigger, you have got to recruit people and build up the team, outsource the workload, and try to invest your time and funding in a smart way.
BCAS: You chose to return to China and join SIBCB in 2011. As we know, by then, graduated with a Ph.D. degree, you had won the Connecticut Stem Cell Seed Award from the State of Connecticut and started your independent research in the USA. Would you let us know what motivated you to take the decision?
CHEN: Prof. LI Lin, then head of SIBCB, took a tour around USA to recruit talents from top universities. He stopped at many universities and States, including the State of Connecticut, and caught my attention – and my husband’s as well. It would be great to take a job via which you can pursue your dream and meanwhile stay in reach of your family: taking this position would mean that I would be working on the same campus as my husband, who is also a key collaborator of my research.