The Tan Kah Kee Young Scientist Award in Chemistry goes to Prof. HUANG Xiaoqiang from Nanjing University, recognizing his pioneering contributions to the development of a novel thiamine-dependent photobiocatalytic system. Through protein directed evolution and interdisciplinary innovation, HUANG’s work has expanded the boundaries of biocatalysis and offered a solution to one of the major bottlenecks in chemical synthesis: precise stereochemical control over highly reactive, free prochiral radicals.
For decades, achieving precise control over highly reactive radicals has been a major challenge in synthetic chemistry. Nature itself evades the chemical chaos of free radicals: Natural enzymes prefer safe, predictable two-electron mechanisms and steer clear of unpredictable single-electron radical pathways. While enzymes are one of nature’s most precise molecular architects, free radicals are like untamed beasts—highly reactive and hard to control, yet invaluable for building complex chemical structures.
Now, a synergy of light-driven chemistry and enzymatic precision has achieved the seemingly impossible feat. Researchers have successfully reprogrammed a natural enzyme to herd these wild free radicals, meticulously assembling highly complex pharmaceutical building blocks.
The Chaos of Chirality and Free Radicals
To understand the sheer magnitude of this achievement, one must first grasp the concepts of chirality and radical.
Many biological molecules exist in two mirror-image forms—much like your left and right hands. While they share an identical composition, their biological effects can differ drastically. One version of a drug might cure a disease, while its evil twin could cause devastating side effects. Consequently, chemists spend their careers devising ways to selectively synthesize just one specific “hand” of a molecule, striving to raise the ratio of the preferred chiral type in the products—a metric called enantiomeric excess.
As for free radicals, these open-shell species are like chemical toddlers on a sugar rush—they react violently and unpredictably with almost anything they touch. Controlling them to produce a single, specific chiral orientation is a monumental challenge.
Tamed by Light and Directed Evolution
Led by HUANG Xiaoqiang at Nanjing University, a team of scientists decided to rewrite these biological rules. They focused on a family of proteins known as thiamine diphosphate-dependent enzymes, specifically one named benzaldehyde lyase (PfBAL) derived from the bacterium Pseudomonas fluorescens. In its natural state, this enzyme safely stitches molecules together without ever touching free radicals. To fundamentally change its diet, the researchers employed a strategy called photobiocatalysis, which uses visible light to steer enzymatic reactions.
Imagine the enzyme as a highly specialized, microscopic factory. HUANG’s team essentially installed a solar panel on the roof and completely redesigned the factory floor. By introducing a light-absorbing photocatalyst—like a ruthenium complex or the organic dye eosin Y—they used flashes of visible light to strip a single electron from a precursor molecule. This precisely timed zap of energy intentionally generated the dreaded enzymatic radical right inside the enzyme’s active site and free radical in the solution, thereby leading to new-to-nature radical-radical cross-coupling.
In their initial breakthrough, the team engineered the enzyme to perform radical acylation. They mutated specific amino acids within the enzyme’s pocket—swapping out atomic components at a microscopic level through directed evolution—to create a bespoke mold. When the light-activated radical was formed, this intricately shaped pocket physically forced the highly reactive species to bond with a second molecule in a single orientation. This yielded chiral ketones with up to 97% enantiomeric excess, proving that radicals could indeed be tamed and transformed into specific chiral products. In short, chiral selection is achieved by mutating the enzyme to construct a perfectly shaped atomic funnel. It uses bulky amino acids to physically block unwanted radical orientations, while simultaneously using attractive dispersion forces to pull the intermediates into the exact energetic “sweet spot” required to form a single, specific chiral mirror image.
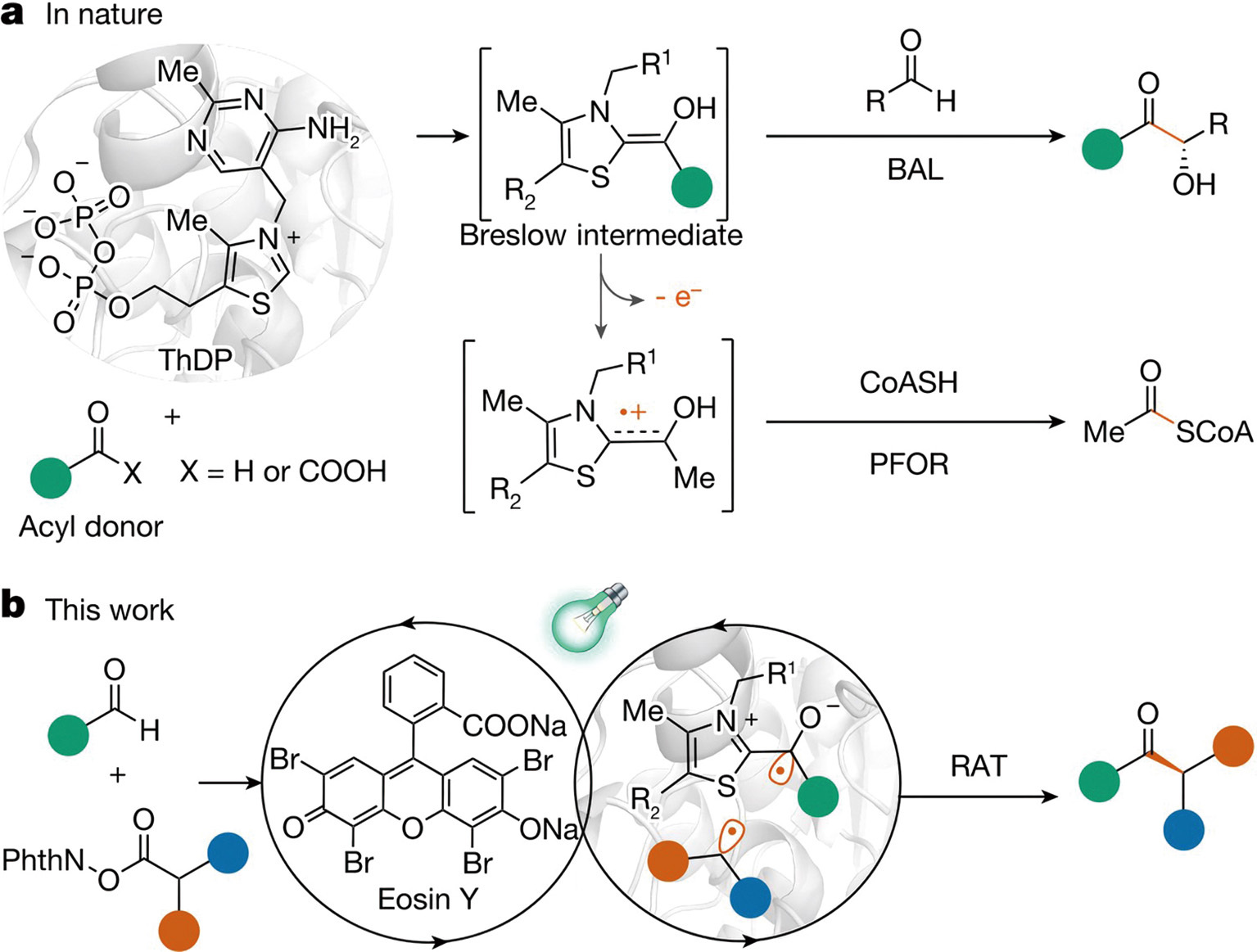
Repurposing a lyase into a Radical Acyl Transferase (RAT). a, The natural baseline: ThDP-dependent enzymes typically build carbon bonds using standard two-electron pathways, with only rare exceptions involving radicals (such as in PFOR catalysis). b, The new breakthrough: A novel dual system combining light (photoredox) and enzymes (biocatalysis) to drive an unnatural, highly precise (enantioselective) radical transfer. (CoASH: coenzyme A; PhthN: phthalimide). (Graphic: Xu et al., 2024)
Having proven the viability of this dual photoredox and biocatalytic system, the next step was to evaluate its versatility. To determine its broad applicability, the researchers introduced diverse aldehydes and carboxylic acid derivatives (N-(acyloxy)phthalimides) into the system. The team demonstrated that the system is exceptionally robust and general. It seamlessly accommodates a wide array of aromatic aldehydes bearing electron-donating or electron-withdrawing groups, as well as heteroaryl aldehydes (such as pyridine and indole). Remarkably, it enables direct and precise structural modifications on commercially available anti-inflammatory drugs (like loxoprofen and flurbiprofen). Furthermore, this reaction can be scaled up to a preparative 1-mmol level without any loss in stereoselectivity.
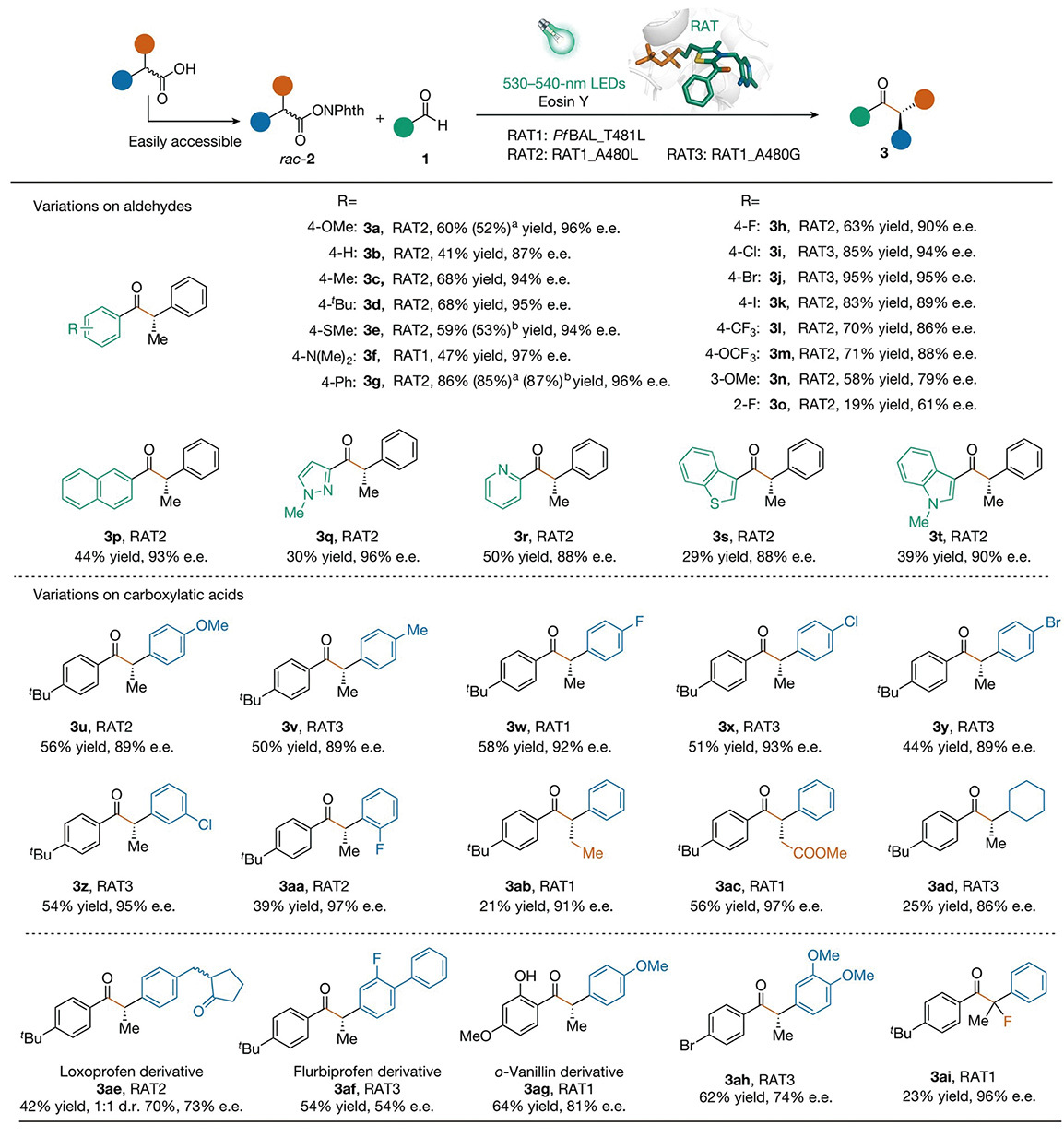
Broad substrate scope of the photobiocatalytic system. This dual photoredox and biocatalytic method is highly versatile and applicable to a wide substrate scope. It successfully accommodates a diverse array of variations on both aldehydes (top) and easily accessible carboxylic acids (bottom), reliably producing a rich library of chiral ketones with high yields and excellent enantioselectivities. (Graphic: Xu et al., 2024)
The “Triple-Radical Sorting” Milestone
In a stunning follow-up, the team tackled a problem that has haunted biochemists for decades: triple-radical sorting. Multicomponent reactions, where three or more raw materials are thrown into a flask to spontaneously form a complex product, are chemical goldmines because they rapidly build complexity. Attempting this with three distinct free radicals, however, almost always results in a chaotic, useless sludge. The mutated enzyme, dubbed a three-component radical enzyme, somehow managed the chaos.
It combined an aldehyde, an alpha-bromo-carbonyl, and an alkene. The photocatalyst absorbed blue light and kicked off an electron-transfer relay that generated distinct radicals. The enzyme’s meticulously evolved pocket—specifically a variant featuring five crucial amino acid changes like T481L and A480G—acted like a microscopic traffic cop. It directed the rapid, sequential collision of these three chaotic components, effectively preventing them from reacting randomly with one another. The raw data proved the sheer elegance of this traffic control. Out of 33 different chemical variations tested, an astounding total of 24 achieved an enantiomeric excess of 97 percent or higher.
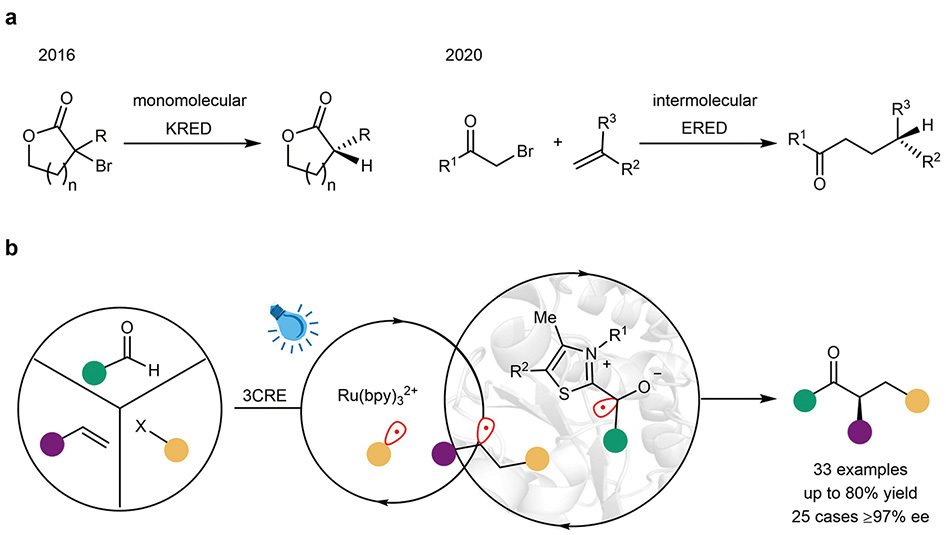
Evolution of photobiocatalysis for triple-radical sorting. a, Previous milestones: Earlier photobiocatalytic reactions were mainly limited to one- or two-component transformations. b, This work: A photobiocatalytic three-component radical cross-coupling system successfully tackles the core challenges of overriding native enzyme activity and managing three distinct radicals with high chemo- and enantioselectivity. (Graphic: Xing et al., 2025)
How exactly does the enzyme prevent the radicals from recombining into useless byproducts? The researchers used supercomputers to run molecular dynamics simulations and quantum mechanics calculations, which allowed them to peer directly into the atomic ballet taking place within the enzyme.
They discovered that specific amino acids act as physical and chemical guardrails. For instance, an amino acid at position 113—mutated to a histidine—functions as a chemical base to strip away a proton (H+), smoothly converting one radical intermediate into another with an incredibly low energy barrier. Meanwhile, the steric hindrance—the sheer physical bulkiness—of a leucine amino acid at position 481 physically blocks the radical from approaching from the wrong direction.
Forging New Synthetic Pathways
The broader implications of this synergistic photobiocatalysis are profound. 2018 Nobel laureate Frances Arnold described the work as one of the most exciting new directions for non-natural enzymatic transformations, underscoring its paradigm-shifting nature. The international chemistry community hailed it as a remarkable achievement, recognizing that this hybrid approach solves a critical bottleneck in the green and selective synthesis of molecules.
This breakthrough has garnered worldwide acclaim. The chemical journal C&EN specifically highlighted the work, quoting Prof. Fasan, who called it “a beautiful piece of work and a remarkable achievement in biocatalysis”. Furthermore, some leading experts have recognized its significance in top-tier journals: Prof. XIAO Wenjing’s team praised it in Angewandte Chemie as a new pathway for the highly efficient and precise synthesis of complex molecules, while Prof. LIU Xinyuan described it in Science as a representative advancement in the field of radical asymmetry.
By marrying light-induced free radicals with the directed evolution of biological enzymes, scientists have crossed a fundamental boundary. Biocatalysis is no longer limited to the reactions that nature painstakingly evolved. Scientists can now forge their own synthetic pathways, crafting molecules of extraordinary complexity with the simple flip of a light switch.
Reference
Xing, Z., Liu, F., Feng, J., Yu, L., Wu, Z., Zhao, B., . . . Huang, X. (2025). Synergistic photobiocatalysis for enantioselective triple-radical sorting. Nature, 637(8048), 1118–1123. doi: 10.1038/s41586-024-08399-5
Xu, Y., Chen, H., Yu, L., Peng, X., Zhang, J., Xing, Z., . . . Huang, X. (2024). A light-driven enzymatic enantioselective radical acylation. Nature, 625(7993), 74–78. doi: 10.1038/s41586-023-06822-x