The Tan Kah Kee Young Scientist Award in Life Sciences recognizes Prof. LIU Xing from the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences for his groundbreaking research into host anti-infection immunity. By tracing the molecular pathways of cellular defense, LIU’s work bridges fundamental immunology and translational medicine. His studies revealed how skin cells detect invading pathogens and initiate pyroptosis—a highly regulated form of inflammatory cell death. Furthermore, his research traced how this localized defense mechanism can escalate into deadly systemic sepsis and identified a repurposed pharmaceutical intervention to halt the resulting cytokine storms. Together, these discoveries offer a comprehensive framework of how the human body balances targeted cellular sacrifice with the risk of systemic self-destruction.
To understand how this delicate balance operates in practice, LIU’s team began their investigation at the primary front line of host defense: the skin, where an arms race between bacteria and human cells plays out at the microscopic level.
Turning the Pathogen’s Weapon against Itself
Group A Streptococcus (GAS) is a virulent bacterium responsible for severe skin infections, utilizing a secreted protease called SpeB to invade host tissues. LIU’s first major investigation revealed a brilliant evolutionary countermeasure: human skin cells use a dormant protein called GSDMA as a tripwire.
To understand how this weapon worked, the researchers first observed live mice. They found that normal GAS caused nasty, localized skin ulcers packed with immune cells. However, when they infected mice with a mutant GAS lacking the SpeB enzyme, the bacteria caused very little skin damage but rapidly triggered a lethal, full-body infection.
Notably, the team discovered that SpeB is harmless outside the cell. It was only when they used a mild electric shock (electroporation) to force SpeB inside isolated skin cells that the cells rapidly swelled and burst (pyroptosis). To find out what SpeB was attacking inside the cell, they ran a genome-wide CRISPR screen, deleting thousands of genes individually across a batch of cells. When they shocked SpeB into these mutated cells, the only survivors were those missing the GSDMA gene.
In in vitro assays, the researchers demonstrated that SpeB directly cleaves GSDMA at a precise location. This targeted cut liberates the N-terminal fragment of GSDMA, which aggressively seeks out acidic lipids (like phosphatidylserine and cardiolipin) on the inner cell membrane. Once anchored, multiple fragments oligomerize to punch physical pores through the membrane structure, effectively sealing the infected cell’s explosive fate.
Membrane pore formation during pyroptosis. (Image generated with AI)
To prove this mattered for survival, they engineered mice lacking the Gsdma1 gene. When infected with GAS, these mice couldn’t trigger the localized cellular explosions. Without this immune alarm, the bacteria escaped the skin, invaded the blood and organs, and killed over half the mice within 5 days.
These discoveries proved that our bodies have evolved a brilliant way to turn a bacterium’s own weapon against it. By acting as both a sensor for the SpeB enzyme and an executioner that blows up the cell, GSDMA ensures that the invading GAS triggers a massive local immune response, trapping the bacteria in the skin before it can cause a lethal whole-body infection.
The Lysosome as an Immune Command Center
While skin cells have evolved a brilliant tripwire against GAS infection, the evolutionary arms race never stops. Pathogens continually evolve more insidious ways to evade or dismantle host defenses. Previous studies showed that another bacterium, Yersinia, injects a weapon called YopJ to disable the host’s primary immune alarm, the TAK1 kinase. The host cell senses this sabotage and triggers a backup self-destruct sequence involving the executioner proteins RIPK1 and caspase-8. The question was: How does a blinded cell know to flip this backup switch?
Once again, the team turned to a massive CRISPR screen. They treated thousands of mutated mouse immune cells with a chemical combo (LPS + 5z7) that mimics Yersinia’s YopJ sabotage. When they sequenced the DNA of the rare surviving cells, they found a massive surprise: the missing genes belonged to the Rag-Ragulator complex. This was shocking because the Rag-Ragulator complex is famously known as a metabolic “fuel gauge” that sits on the lysosome (the cell’s recycling center) to monitor environmental cues, such as the availability of nutrients and amino acids.
By tracking the physical location of the cell’s proteins, the researchers found that during a Yersinia infection, the executioner proteins physically rush to the surface of the lysosome to bind to the Rag-Ragulator complex. To prove the lysosome was a mandatory “launchpad” for pyroptosis, they genetically engineered the Ragulator so it floated freely in the cell instead of anchoring to the lysosome. When unanchored, the self-destruct sequence completely failed.
When they used drugs like rapamycin to block the Rag-Ragulator’s normal metabolic day job (turning on a major metabolic engine called mTORC1, which tells the cell to grow), the cells still exploded. This proved that the Rag-Ragulator complex was effectively moonlighting—acting as an independent, physical scaffold for the immune system, entirely separate from its usual metabolic job.
Finally, they looked at how the Rag-Ragulator complex pulls the trigger. They found that the complex utilizes a motor-like protein called RagC. They also found that RagC must be in a specific, active state (bound to a molecule called GDP) to work. When they locked RagC in an inactive state, the caspase-8 protein couldn’t activate, and the cell survived the Yersinia infection without exploding.
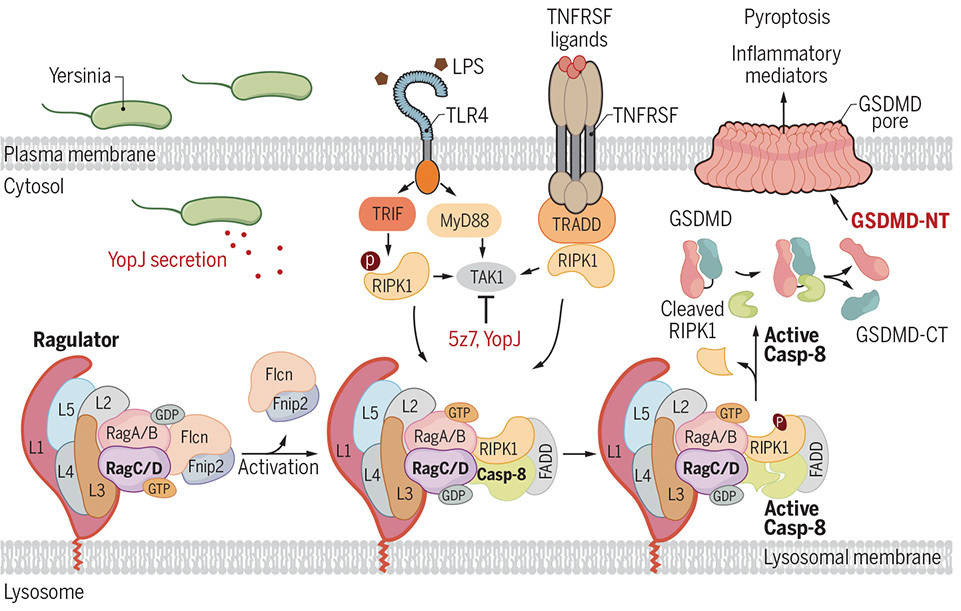
The Rag-Ragulator Backup Alarm. When Yersinia’s YopJ toxin disables the host’s TAK1 kinase, the cell relies on the lysosomal Rag-Ragulator complex to trigger a backup self-destruct sequence. (Graphic: Zheng et al., 2021)
It seems that lysosomes aren’t just for recycling cellular trash; they are active immune command centers. The cell brilliantly uses its metabolic nutrient sensor (Rag-Ragulator) as a physical launchpad. When Yersinia bacteria try to quietly sabotage the host’s primary immune alarms, the Rag-Ragulator complex senses the danger, recruits the executioner proteins RIPK1 and caspase-8 to the lysosome, and blows up the cell through pyroptosis to alert the rest of the body.
The Paradox of Sepsis and Mitochondrial Amplification
While pyroptosis is an effective localized defense, it becomes a systemic nightmare if the infection reaches the bloodstream. Widespread pyroptosis initiates an uncontrolled cytokine storm, culminating in sepsis—a lethal cascade that destroys blood vessels and causes multi-organ failure.
To understand this catastrophic amplification, LIU’s team investigated the intracellular targets of another executioner protein, GSDMD. The team found that during severe infections, cleaved GSDMD does not immediately target the outer cell membrane. Instead, it turns its deadly attention inward, specifically seeking out cardiolipin on the cell’s mitochondria.
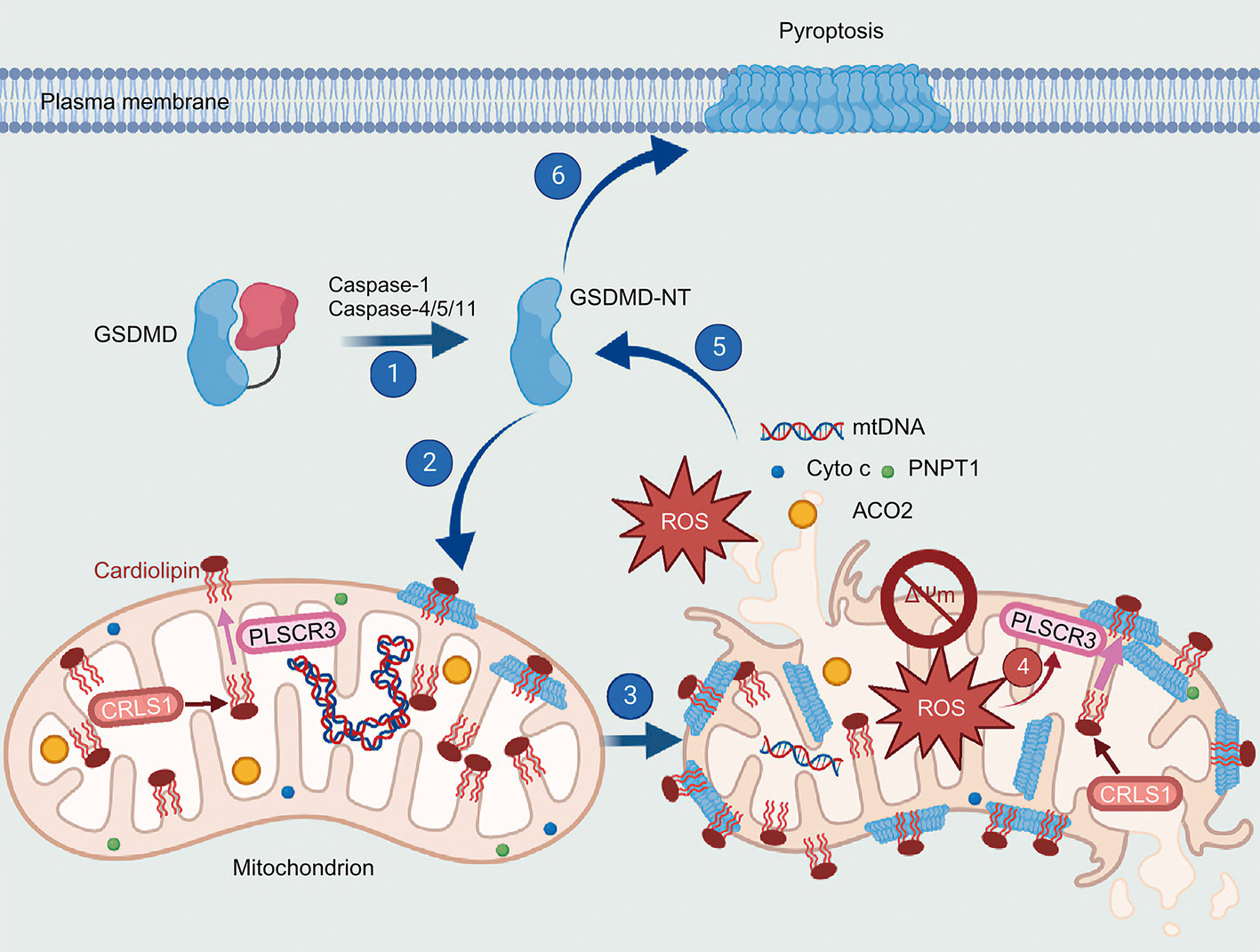
Mitochondrial damage is a "point of no return" that is critical for cell death and inflammation. (Graphic: Miao et al., 2023)
By punching irreversible holes through the mitochondrial membranes, GSDMD causes a catastrophic power plant meltdown. The mitochondria lose their ability to perform oxidative phosphorylation and begin leaking highly toxic reactive oxygen species (ROS) and fragments of mitochondrial DNA (mtDNA) straight into the cellular fluid. These misplaced, toxic byproducts act like gasoline on a fire, supercharging the cell’s internal inflammatory sensors to produce even more executioner enzymes and driving the severe pathology of sepsis.
Repurposing Therapeutics to Halt the Cytokine Storm
Developing a new drug to halt sepsis takes decades—time that patients in septic shock do not have. Recognizing this, LIU and his collaborators embarked on a massive, high-throughput screening effort of thousands of small molecules. They struck gold with Disulfiram, commonly recognized by its brand name Antabuse, a drug safely used for over half a century to treat alcohol addiction.
While it may seem paradoxical to silence the body’s protective inflammatory “alarm system,” doing so with Disulfiram is a lifesaving intervention during the systemic crisis of sepsis. In a localized infection, this alarm is crucial; a few immune cells detect the invading bacteria and undergo a fiery explosion called pyroptosis, which releases inflammatory signals to call for backup and trap the invaders in that specific spot.
However, the rules change entirely during sepsis, a condition where the infection escapes local control and bacterial toxins spread throughout the entire bloodstream. Instead of a localized response, millions of immune cells across the body simultaneously detect the bacteria using their TLR4/MD-2 receptors and undergo pyroptosis, dumping massive amounts of toxic inflammatory cytokines into the blood.
This uncontrolled chain reaction, known as a cytokine storm, means the patient’s own panicked immune system—rather than just the bacteria—becomes the primary threat, driving severe pathology that destroys blood vessels, drops blood pressure to fatal levels, and causes multiple organs to shut down.
In this scenario, Disulfiram acts as a necessary emergency brake by cutting the communication lines (blinding the TLR4/MD-2 receptors) and jamming the executioner’s weapon (blocking GSDMD pore formation). By forcibly preventing the immune cells from exploding, Disulfiram silences the deadly immune overreaction and prevents organ failure, ultimately buying the patient vital time to survive while doctors administer powerful intravenous antibiotics to clear the underlying infection.
Disulfiram: A repurposed double-whammy therapeutic. (Graphic: Ben Mills/Wikipedia)
The impact of this translational research is profound, bridging the gap between fundamental discoveries and tangible drug development. By decoding the intricate dynamics of proteins like GSDMA, GSDMD, SpeB, and the Rag-Ragulator complex, scientists have precisely mapped a specific battlefield of human immunity. This breakthrough has earned Dr. LIU global acclaims, including recognition as a top 2% scientist and an Elsevier Highly Cited Researcher, along with prestigious invitations to author reviews in Nature Reviews Molecular Cell Biology and Nature Reviews Drug Discovery.
The story of pyroptosis highlights the delicate balance of cellular sacrifice and survival. It reveals how easily our protective defenses can plunge into fatal self-destruction—but more importantly, it arms humanity with the pharmacological tools to extinguish these deadly biological flames before they consume us.
Reference
Bai, Y., Min, R., Chen, P., Mei, S., Deng, F., Zheng, Z., . . . Liu, X. (2023). Disulfiram blocks inflammatory TLR4 signaling by targeting MD-2. Proc Natl Acad Sci USA, 120(31), e2306399120. doi: 10.1073/pnas.2306399120
Deng, W., Bai, Y., Deng, F., Pan, Y., Mei, S., Zheng, Z., . . . Liu, X. (2022). Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature, 602(7897), 496–502. doi: 10.1038/s41586-021-04384-4
Hu, J. J., Liu, X., Xia, S., Zhang, Z., Zhang, Y., Zhao, J., . . . Wu, H. (2020). FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nature Immunology, 21(7), 736–745. doi: 10.1038/s41590-020-0669-6
Miao, R., Jiang, C., Chang, W. Y., Zhang, H., An, J., Ho, F., . . . Lieberman, J. (2023). Gasdermin D permeabilization of mitochondrial inner and outer membranes accelerates and enhances pyroptosis. Immunity, 56(11), 2523–2541.e2528. doi: 10.1016/j.immuni.2023.10.004
Zheng, Z., Deng, W., Bai, Y., Miao, R., Mei, S., Zhang, Z., . . . Liu, X. (2021). The Lysosomal Rag-Ragulator Complex Licenses RIPK1 and Caspase-8-mediated Pyroptosis by Yersinia. Science, 372(6549). doi: 10.1126/science.abg0269