
Viruses are among the smallest infectious agents on earth, consisting of genetic materials enclosed in a coating shell of capsid protein. They are basically as simplified as small pods of infectious genetic materials; but they are natural killers and could kill in an artful way – for that, you have to give them credit. It is thought that viruses are everywhere on earth – “where there are lives, there are viruses” – and almost every living thing has a virus that infects it.
They can’t do much alone; they are even not qualified as being alive; but they can always find their ways – usually by binding to receptors from the cell surface – to sneak into a host cell and hijack its machinery to multiply. They are sort of like “parasites” that prey on cells. After replicating into huge numbers, they outburst from the infected cell and spread by repetitive invasions into other cells, which finally cause massive infections and make the host feel sick.
They also have different personalities. Some of them are vicious, rampaging from cell to cell like a hurricane and causing the host to die in a short time; others are more tempered, hiding themselves in the host’s genome, biding their time and waiting for a better chance to make its move. Besides, most human viruses attack specific tissues. For example, human flu viruses infect only cells lining the upper respiratory tract; the HIV binds only to certain white blood cells, and poliomyelitis infects only nerve cells.
To fight against viruses, no matter who they are, it is essential to know how exactly the viruses get into the victim cells, or which accomplice receptor(s) are responsible for the viral entry.
In this piece, we are going to learn a particular group of enteroviruses and how they trick their ways into the victim cells.
The group of Enterovirus B (EV-B), named after their transmission-route through the intestine, consists of echovirus, coxsackievirus B, coxsackievirus A9, and other newly identified enteroviruses. EV-B viruses are associated with many severe brain diseases, such as viral encephalitis, viral meningitis, and viral meningo-encephalitis, leading to substantial morbidity and mortality in children. Over the past few years, global EV-B outbreaks have imposed a substantial risk on children. For example, in April 2019, an enterovirus outbreak occurred in the Shunde Hospital of Southern Medical University (China), leading to the death of five newborns.
Up to now, the viruses still hold the upper hand due to our limited knowledge about how EV-B viruses cross the blood-brain barrier to infect brain tissues. To win this war, scientists need to find out what are the key accomplices that these viruses hijack to sneak into cells and transmit across the blood-brain barrier.
To successfully penetrate into cells and multiply, EV-B viruses need to firstly attach to certain receptors at the cell membrane and sneak into cells via endocytosis, a process of engulfing receptor-bound molecules or particles. But cell entry alone does not guarantee a successful viral propagation and infection. The entered virus needs to take off its coating shell of capsid protein in a process known as uncoating, to release its infectious genetic materials that encode the instructions to assemble new infectious viral particles.
Former studies have identified several receptors (such as CD55, a membrane receptor) that mediate the cell attachment of different EV-B viruses. However, none of them can induce the viral particles to undergo a dramatic structural change that leads to virus uncoating, a process involving the removal of certain capsid proteins and release of the viral genome, which is prerequisite for the successful viral entry. Up to now, only one receptor is confirmed to decoat coxsackievirus B virus, one type of EV-B viruses. The uncoating receptor(s) for most EV-B viruses remained elusive until a recent report published in the journal of Cell. The new finding was made by a team led by Dr. George F. Gao from the CAS Institute of Microbiology and their collaborators from Peking University and the Beijing Children’s Hospital.
A Hunt for the Other Accomplice
To identify the other accomplice that acts to decoat the EV-B viruses, scientists performed a clever and efficient “Infection-and-Survival Test”: they constructed a pool of mutant cells each with a different membrane receptor crippled; the ones that happen to have the key accomplice receptors crippled would deny a potential viral entry, hence would block the viral infection and survive the test.
First, the researchers sought to construct the pool of mutant cells with CRISPR-Cas9, a widely used gene-editing tool inspired by the bacterial immune system. Bacteria take DNA snippets from the invading viruses and store these alien DNA segments known as CRISPR arrays into their genome, keeping them as sort of “criminal records”. To defense themselves, the bacteria produce RNA molecules from these documented CRISPR arrays and loaded them individually onto the Cas9 enzyme to form an array of different defense machines. Once a viral attack occurs, these machines can quickly act to target and destroy the invaders’ genomes, guided by the distinctive RNA codes in its “arsenal.”
Similarly, researchers created customized CRISPR arrays using a pool of single guide RNAs (sgRNA) that can target all human membrane protein-encoding genes. Unlike the bacterial CRISPR arrays that exist within one single cell, each sgRNA molecule from the pool is set to be introduced into a different cell to cripple its target gene. As a result, a pool of mutant cells was created and ready for the viral test.
Then, they exposed these mutant cells to a strain of highly pathogenic Echo 6 virus. Cells that carry the functional accomplice receptors would be infected and killed by the viruses, while the ones that happen to carry a crippled version of accomplice receptors would withstand the infection test and multiply to dominate the mutant pool. By comparing the differences of sgRNA genes between the tested cells (i.e., the survivals) and the untested cells, they discovered three genes that show higher abundances within the tested group: one encodes CD55; the other two encode the two subunits of the neonatal Fc receptor (FcRn).
CD55 had been formerly identified as an attachment receptor that mediates the cell attachment of Echo 6 virus and many other types of EV-B viruses. But, CD55 can’t decoat any of them. The outcome of CD55 from the test confirmed the effectiveness of the test in locating the accomplices. Notably, CD55-crippled cells were still vulnerable to echovirus infection, though at a reduced rate. This observation suggested the existence of “other receptor(s)” that also mediate viral attachment. The identity of such “other receptor(s)” that also mediate Echo 6 attachment will be revealed in the following chapter.
To verify FcRn’s role in echovirus infection, they infected the FcRn-crippled cells with Echo 6 virus and found that the dysfunction of FcRn completely denies Echo 6 virus infection. Hence, it seems that FcRn is the other accomplice receptor that is exploited by the echovirus to decoat itself and to release its infectious genome.
Interrogation and Verdict
To confirm if FcRn is indeed the other accomplice that mediates Echo 6 uncoating, they interrogated FcRn in many ways.
First, they sought to verify if FcRn indeed directly binds to EV-B viruses. Because a direct binding to the virus is demanded for FcRn to act as the uncoating receptor. Molecular interaction analyses showed that the extracellular domain of human FcRn protein binds to Echo 6 with a high affinity, just like CD55.
Next, they sought to probe the potential function of FcRn in EV-B entry. Unlike CD55 that is wholehearted to mediate viral attachment, they found that the dysfunction of FcRn had a very limited effect on Echo 6 binding to the cell surface. This suggests that FcRn plays its essential role after viral attachment. However, when CD55 is out of function, FcRn alone can also suffice viral entry, though at a reduced rate. This explains why the CD55-crippled cells were still vulnerable to echovirus infection, as mentioned above. Hence, FcRn is the answer to the riddle of who could be the “other receptor(s)” that also medicate the cell attachment of Echo 6 virus.
To illustrate the essential role of FcRn in EV-B entry, researchers resorted to cryogenic electron microscopy (Cryo-EM), a technique widely used for snapshotting and 3D-reconstructing molecular structures.
Considering there are two locations vital for the virus entry, the cell membrane and the endosome, researchers snapshotted Echo 6 virus alone and in complex with CD55 or FcRn under both neutral (pH 7.4) and acidic (pH 5.5) conditions, where the neutral pH mimics the condition at the cell membrane and the acidic pH mimics the endosomal environment. They found that CD55-bound viral particles show no sign of structural change under either pH condition; nor do the FcRn-bound particles at neutral pH. However, the acidic pH caused a dramatic structural change to FcRn-bound viral particles, in particular the collapse of a pocket in the coating shell. The collapsed pocket pushes out the “pocket factor”, a lipid molecule that functionally mimics suture that keeps the viral particle intact. The pocket factor release further triggers a structural change and destabilizes the viral particle, which finally leads to viral uncoating and the resultant release of viral genome. So, the structural evidence indicates that FcRn mediates virus binding, ‘‘pocket factor’’ release, and capsid uncoating, whereas CD55 solely mediates the viral attachment.
To further back up their discovery and see it for sure, they simulated the uncoating event by an in vitro experiment, where Echo 6 full particles were incubated with FcRn-decorated liposomes at acidic condition to mimic what happens within endosome during viral entry. As they expected, a considerable proportion of empty particles were clearly distinguishable from the full particles after a short incubation.
Could FcRn be exploited by other types of EV-B viruses? To answer that, scientists individually tested the FcRn-crippled cells with 11 different types of echoviruses and two other EV-B viruses, which together feature the major EV-B serotypes – here, one serotype means a group of related viruses distinguished by a common set of antigens. The results showed that the FcRn-crippled cells are not infected by any selected virus. They thus proposed that FcRn is the key accomplice receptor that decoats most EV-B viruses during cell entry.
Based on the converging evidence, the researchers proposed a dual-receptor model for enterovirus entry. The viral particle first binds to the attachment receptor CD55 at the cell membrane, and triggers the endocytosis, a process of engulfing receptor-bound molecules or particles. As a result, the enterovirus sneaks into the endosome, where the FcRn receptor somehow takes over the CD55-bound viral particles. The acidic pH within the endosome prompts the extrusion of ‘‘pocket factor’’ after FcRn binding, resulting in viral uncoating and genome release, which are critical to initiate the viral replication cycle. In addition, viral particles could also be probably captured by FcRn alone at the cell membrane or by FcRn and CD55 together, as FcRn binds to viral particles at both neutral and acidic pH, and further induces endocytosis and uncoating in the endosome.
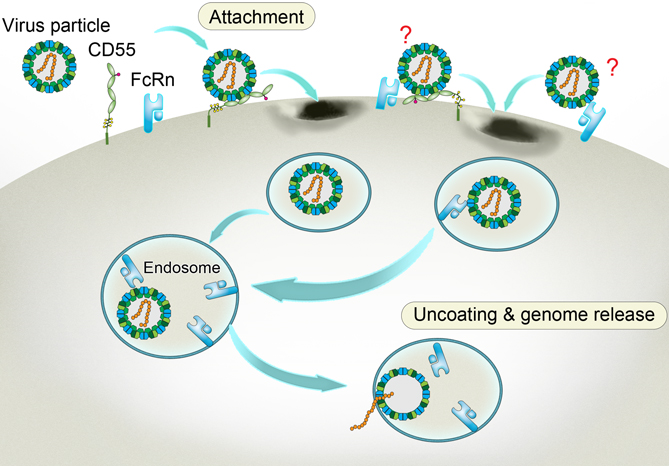
A way to get inside aided by the accomplice receptors. Enterovirus binds to CD55 at the cell membrane and sneaks into the inward-budding endosome, where the viral particle binds to another receptor FcRn. Triggered by the acidic pH within the endosome, the FcRn-bound particle undergoes a structural change. As a result, the viral particle decoats itself and releases its infectious genome that heralds the propagation of the virus and the doom of the cell. The two undefined receptor binding modes on the viral surface are labeled with question marks. (Credit: Dr. George F. Gao from the CAS Institute of Microbiology)
A New Chance to Fight Back
Normally, FcRn helps transporting maternal immunoglobulin G (IgG) across the placenta to the fetus and across the gut to the bloodstream of the newborn. The new finding indicates otherwise that FcRn can be exploited and hijacked as a key accomplice by most EV-B viruses to sneak into cells and release their infectious genomes. Besides, FcRn exists abundantly in the gut and at the blood-brain barrier. This is in accordance with the tissue tropism of EV-B viruses, as the gut is the major target for EV-B viruses, where they transmit to other organs such as the brain and cause severe infections. As the accomplice, FcRn may also help EV-B viruses cross both the blood-brain barrier and the blood-placenta barrier. Hence, FcRn is a high-value target involved in EV-B-induced infections and is worth a further look under physiological conditions.
Thus far, there is no approved drug or vaccine specifically against EV-B infection due to our limited knowledge and lack of animal models. However, the identification of FcRn as the key receptor for most EV-B viruses opens a new path to meet these medical needs. On the one hand, this newly-identified key receptor provides a target for medical interferences. On the other, it also provides a possibility to construct animal models to study how EV-B viruses cause many severe brain infections, as well as to develop antiviral drugs and vaccines. In particular, scientists could genetically modify rodents to carry human FcRn to enable their echovirus susceptibility. Thus, these modified animals present a valuable model for EV-B virus study.
The identification of FcRn as a universal receptor for echovirus to sneak into cells may shed lights on developing echovirus-based oncolytic therapeutics. Echoviruses exploit key receptors to intrude into cells, replicate themselves and burst out, which finally causes the death of victim cells. Many of these key receptors happen to be overexpressed in cancer cells. Besides, echoviruses have preference to infect certain tissues, such as gut and brain cells. Thus, echoviruses, native or genetically modified, could be used as “hired guns” to specifically kill cancer cells.
Reference
X. Zhao et al., Human Neonatal Fc Receptor Is the Cellular Uncoating Receptor for Enterovirus B. Cell 177, 1553 (Published: May 16, 2019). doi: 10.1016/j.cell.2019.04.035.